


JE M'ABONNE DÈS AUJOURD'HUI
et j'accède à plus de contenu
| ISO 690 | Schwitzgebel, V., Dirlewanger, M., Fatio, S., Meier, C., A., Traitement par hormone de croissance : phase de transition de l’enfance à l’âge adulte, Rev Med Suisse, 2005/006 (Vol.1), p. 426–430. DOI: 10.53738/REVMED.2005.1.6.0426 URL: https://www.revmed.ch/revue-medicale-suisse/2005/revue-medicale-suisse-6/traitement-par-hormone-de-croissance-phase-de-transition-de-l-enfance-a-l-age-adulte |
|---|---|
| MLA | Schwitzgebel, V., et al. Traitement par hormone de croissance : phase de transition de l’enfance à l’âge adulte, Rev Med Suisse, Vol. 1, no. 006, 2005, pp. 426–430. |
| APA | Schwitzgebel, V., Dirlewanger, M., Fatio, S., Meier, C., A. (2005), Traitement par hormone de croissance : phase de transition de l’enfance à l’âge adulte, Rev Med Suisse, 1, no. 006, 426–430. https://doi.org/10.53738/REVMED.2005.1.6.0426 |
| NLM | Schwitzgebel, V., et al.Traitement par hormone de croissance : phase de transition de l’enfance à l’âge adulte. Rev Med Suisse. 2005; 1 (006): 426–430. |
| DOI | https://doi.org/10.53738/REVMED.2005.1.6.0426 |
| Exporter la citation | Zotero (.ris) EndNote (.enw) |
The diagnosis of GH deficiency is difficult to establish : clinical, radiological and hormonal data are combined to suspect the disease. GH stimulation tests are an essential part of the evaluation, although the cut-off values are determined arbitrarily. There are different stimulation tests. Their use depends on the patient’s age. Once the diagnosis is ascertained, the treatment is started and maintened until the end of statural growth. The persistence of GH deficiency needs to be confirmed during the transition phase. If required, GH treatment can be continued until the achievement of peak bone mass. Thereafter the benefit of continuing GH treatment are mainly related to the quality of life. The long term effects on cardiovascular morbidity/mortality are not demonstrated.
Le déficit en hormone de croissance (GH) est difficile à diagnostiquer. L’ensemble des données cliniques, radiologiques et hormonales le font suspecter. Les tests de stimulation de la GH font partie de l’évaluation, leurs valeurs seuils ont été établies arbitrairement. Il existe de nombreux tests, dont l’utilisation est différente en fonction de l’âge. Une fois le diagnostic établi par deux tests, le traitement est poursuivi jusqu’à la fin de la croissance. La phase de transition est caractérisée par la nécessité de réévaluer la persistance du déficit en GH. Dans ce cas, le traitement peut être poursuivi quelques années afin de permettre l’obtention du pic de masse osseuse. A l’âge adulte, d’autres bénéfices semblent exister. L’impact du traitement sur la morbidité/mortalité est inconnu.
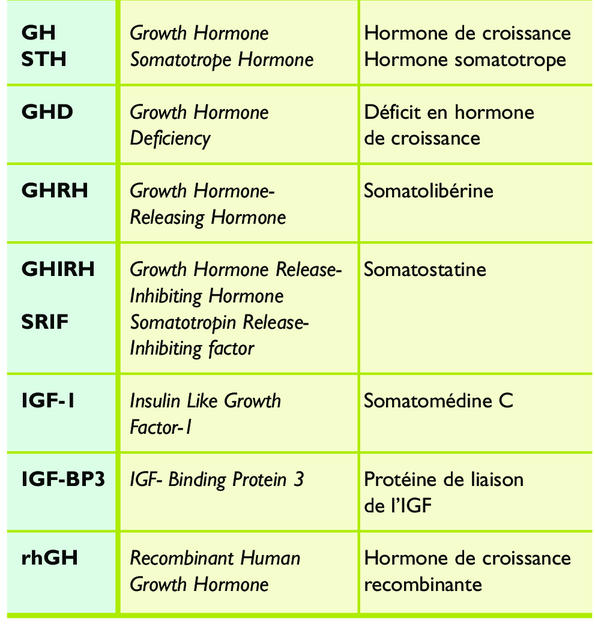
Tableau synonymes et abréviations
La période de transition est définie comme la période survenant après l’accomplissement de la croissance linéaire, mais avant l’atteinte de la maturité somatique et psychologique complète.1 Cette période est capitale dans le suivi d’un enfant/adolescent tout particulièrement en cas d’atteinte endocrinienne. Beaucoup de changements surviennent dans de multiples systèmes durant cette phase, tout particulièrement au niveau de l’axe somatotrope et de la reproduction.
L’hormone de croissance (GH) n’influence pas seulement la croissance, mais également la composition corporelle, la densité osseuse, le bien-être pour ne citer que les principaux. L’approche initiale de la substitution par GH ne s’intéressait qu’à la croissance et se terminait une fois la taille adulte atteinte. Mais le suivi des enfants avec déficit en GH (GHD) a révélé la nécessité de définir les facteurs permettant de prédire la persistance d’un GHD à l’âge adulte et la nécessité de poursuivre le traitement.
Le but de cette revue est de revoir la physiologie de l’axe somatotrope, l’étiologie du déficit en GH chez l’enfant, son traitement avec ses bénéfices et complications à long terme. Nous discuterons également des bénéfices du traitement chez l’adolescent ou le jeune adulte présentant la persistance d’un déficit en GH.
Schéma simplifié de l’axe somatotrope
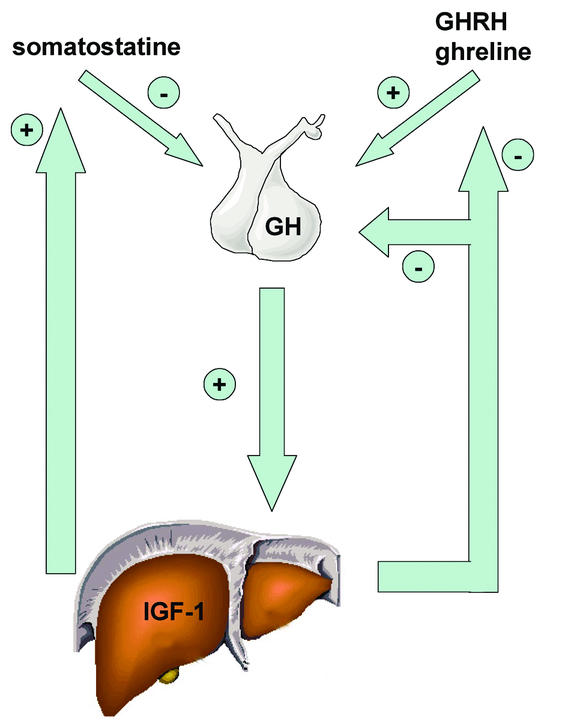
L’hormone de croissance (GH) est sécrétée par l’antéhypophyse après stimulation par la GHRH (GH releasing hormone). Elle est inhibée par la somatostatine. Le site de production de la GHRH est le noyau arqué de l’hypothalamus, alors que les sites de synthèse de la somatostatine sont les noyaux paraventriculaires. La sécrétion de ces deux hormones hypothalamiques est régulée par un réseau neuronal complexe. Une grande variété de neurotransmetteurs (adrénergiques, dopaminergiques, sérotoninergiques et cholinergiques) interagit avec les récepteurs spécifiques, exerçant un effet agoniste ou antagoniste sur les sites de production de ces peptides hypothalamiques. La modification du niveau de sécrétion de GHRH et/ou somatostatine va entraîner une modification de la sécrétion pituitaire de GH. Certains facteurs, comme l’IGF-1 (Insulin-Like Growth Factor-1), la ghreline (un peptide d’origine gastrique), ou les acides gras libres agissent directement sur la sécrétion de GH.2 Par un rétrocontrôle négatif la GH va induire la sécrétion de la somatostatine.
La GH est une protéine d’environ 190 acides aminés, circulant sous une forme de 20 et 22 kD, qui peut stimuler la croissance de nombreux tissus en particulier le cartilage de croissance. Elle augmente la synthèse des protéines et diminue leur catabolisme, elle stimule la lipolyse et induit une résistance à l’insuline.3 Au niveau périphérique, sous l’effet de la GH, le foie produit une protéine, l’IGF-1 (anciennement appelée somatomédine C) qui, en se liant à des protéines de liaison, dont l’IGF-BP3 (IGF-binding protein 3) participe à la stimulation de la prolifération des chondrocytes et la croissance épiphysaire. L’IGF- BP3 est également sous l’influence de la GH.
Le diagnostic du déficit en GH (GHD) est fait par l’ensemble des données auxologiques, cliniques, génétiques, radiologiques, métaboliques et hormonales. Une évaluation fine est effectuée selon les paramètres de croissance (taille inférieure à - 2DS, vitesse de croissance inférieure à - 1DS, infléchissement du chenal de croissance), l’examen clinique (répartition du tissu adipeux, recherche d’anomalies neurologiques) et des données radiologiques (évaluation de la maturation osseuse par une radiographie du poignet et de la main gauche), et finalement par la détermination du taux d’IGF-1 et IGF-BP3 et des tests de stimulation de la GH. Les tests de stimulation ont entraîné la nécessité de définir des valeurs seuils définissant la réponse anormale. La réponse de la GH aux divers stimuli varie avec la méthode, le sexe, l’âge et le développement pubertaire, ainsi que la puissance du test. La valeur seuil reste donc arbitraire. La décision de traiter devrait donc toujours dépendre du contexte clinique et l’on devrait résister à la tentation de traiter simplement des variantes de la norme (la FDA par exemple accepte le traitement des petites tailles - 2DS, sans aucune autre anomalie).
Le déficit en GH comprend toute anomalie impliquant une anomalie de synthèse ou de sécrétion de la GH ou un défaut de son action périphérique (tableau 2).
Etiologies GHD

Dans les causes secondaires, il faut évoquer les pathologies tumorales avec ou sans radiothérapie, les lésions vasculaires, les traumatismes. D’autres pathologies peuvent induire une résistance à l’action de la GH (par exemple l’insuffisance rénale chronique).
La prévalence des GHD est de 1:4000 à 1:30000 selon les études.4 Les causes génétiques surviennent généralement de façon sporadique (mutation des gènes Pit-1, Prop-1, HESX 1, LHX 3, LHX 4), mais certaines atteintes sont transmises par un mode récessif ou dominant (mutation du gène codant pour la GH, ou codant pour la GHRH, ou codant pour le récepteur GHRH). Historiquement, le diagnostic de GHD a été établi empiriquement. On avait observé que l’amplitude de la sécrétion de GH suite à un stimulus connu et entraînant normalement une libération de GH dans la circulation était inappropriée chez certains enfants avec une petite taille par rapport à un enfant grandissant normalement. Les divers aspects méthodologiques en ont découlé.
Le diagnostic du déficit en GH reste complexe et controversé, incluant des données cliniques et de laboratoire. Les difficultés viennent de la variabilité des méthodes utilisables, de l’établissement de valeurs seuils des tests de stimulation de la GH, ainsi que de la mauvaise reproductibilité intra-individuelle. Les voies de stimulation utilisables sont multiples, certaines n’étant utilisées qu’en pédiatrie et d’autres uniquement à l’âge adulte. Nous nous contenterons d’évoquer les plus utilisées, pour lesquelles il existe une large expérience sur le plan international.5-7 Ces différents tests, leurs indications, contre-indications, effets indésirables et leur mécanisme d’action sont résumés dans le tableau 3.
Les tests de stimulation de la GH
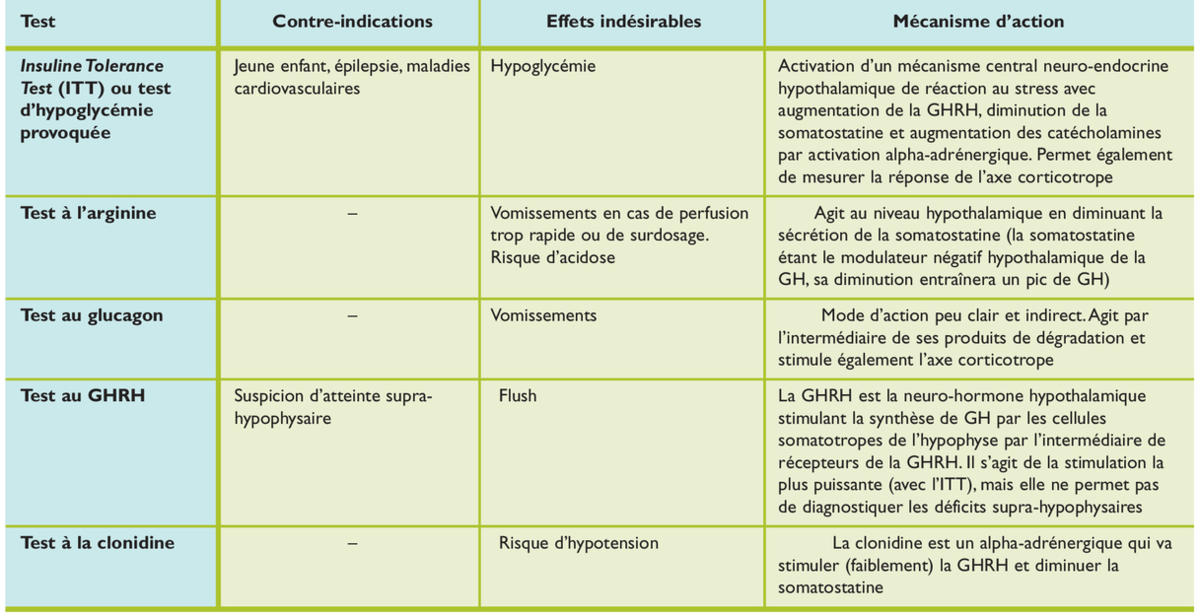
La plupart de ces tests peuvent se faire en combinaison (arginine-insuline, arginine-GHRH) ce qui permet d’agir souvent par deux mécanismes (stimulation de la GHRH et diminution de la somatostatine) et d’obtenir la meilleure élévation possible de la GH.
En raison de leur variabilité intra- et interindividuelle, deux tests de stimulation, effectués à quelques jours d’intervalle, sont nécessaires avant d’affirmer le diagnostic de déficit en GH, en particulier en cas de déficit isolé. En cas d’atteinte de plusieurs axes, en particulier dans le cadre de malformation de l’axe médian, après chirurgie ou radiothérapie, un seul test démontrant le déficit est le plus souvent suffisant.
L’interprétation des tests se fera en fonction de l’âge et des stades pubertaires. En général, on retient les taux < 10 ng/ml, < 5 ng/ml chez l’enfant (déficit partiel et complet) et < 3 ng/ml chez l’adulte, comme étant pathologiques.7 Le chevauchement des valeurs normales et pathologiques induit de nombreux résultats faux positifs ou faux négatifs, raison pour laquelle deux tests sont nécessaires.
La mise sur le marché de GH recombinante humaine (rhGH) en 1986 a nettement élargi l’utilisation du traitement par GH. Les indications reconnues et remboursées par l’assurance sont les déficits partiels ou complets en GH, le syndrome de Turner et l’insuffisance rénale chronique. L’indication et la prise en charge du traitement des retards de croissance intra-utérins sans croissance de rattrapage jusqu’à trois ans et le syndrome de Prader-Willi sont en cours d’évaluation. Le traitement consiste en une injection souscutanée six à sept jours sur sept. La rhGH peut entraîner comme effets indésirables, une intolérance glucidique-diabète (rare), des douleurs osseuses avec risque d’épiphysiolyse, une hypertension intracrânienne bénigne, réversible à l’arrêt du traitement.8
Les enfants avec GHD atteignent donc une taille entrant dans la taille cible familiale, mais inférieure de 2-4%.9,10 La croissance de rattrapage s’effectue principalement durant la première année de traitement. Ce gain durant la première année est le plus prédictif du gain à long terme.11
La GH améliore également la densité osseuse, la force musculaire et plus globalement la qualité de vie.
La crainte de développer un cancer (de novo, une récidive ou une seconde néoplasie après avoir survécu à un premier cancer) sous traitement de GH reste ancrée dans l’esprit de nombreux parents et praticiens. Ceci est lié à la connaissance épidémiologique du lien entre acromégalie (excès en hormone de croissance) et cancer colique, mais aussi aux effets mitogéniques de l’IGF-1 et des autres facteurs de croissance. Cependant, la comparaison avec les acromégalies n’est pas justifiée, le traitement substitutif en GH ne devant pas élever le taux d’IGF-1 au-delà de la norme pour l’âge. Les données existent en ce qui concerne les enfants survivants d’un cancer. La survenue d’une récidive du cancer primaire n’est pas plus élevée chez les enfants survivants traités par GH que chez ceux non traités. Cependant, une augmentation du risque de seconde néoplasie est observée chez les enfants survivants d’une leucémie (RR 3.21) mais un biais important apparaît avec les traitements anticancéreux préalablement reçus (chimiothérapie, radiothérapie, transplantation et immuno-suppression).12-14
Le risque de maladie de Creutzfeld-Jakob a été une réalité lorsque le seul traitement disponible était l’hormone extraite d’hypophyse de cadavre. Depuis 1986, il existe sur le marché l’hormone de croissance fabriquée par génie génétique. La possibilité de transmission de la maladie de Creutzfeld-Jakob est donc nulle actuellement.
Le génie génétique est source de craintes dans le grand public. Il faut se rappeler que cette méthode est utilisée depuis plus de vingt ans pour produire de nombreux médicaments et vaccins, y compris l’insuline. En Suisse, une trentaine de médicaments issus du génie génétique sont disponibles, produits soit dans des bactéries, soit dans des cellules génétiquement modifiées. Ces médicaments présentent l’avantage d’une production illimitée au plan quantitatif, ce que les méthodes traditionnelles ne permettent souvent pas. Mais leur principale qualité réside dans leur pureté, éliminant ainsi le risque de transmission de maladies lié aux méthodes de préparation à partir de tissus humains (VIH en particulier).
La phase de transition entre l’adolescence et le jeune adulte est critique (tableau 4 et figure 2) : la fin de la croissance staturale impose de pratiquer une fenêtre thérapeutique afin de confirmer la persistance du déficit, en particulier en cas de déficit isolé. On retrouve dans la littérature des taux de normalisation de l’axe somatotrope jusqu’à 67% dans ce contexte, tandis que seuls 10% se normalisent lors de GHD associé à une atteinte organique ou une radiothérapie.15 Jusqu’à présent, le traitement était classiquement terminé lorsque la taille adulte était atteinte, soit une vitesse de croissance inférieure à 2 cm par an et une maturation osseuse de plus de 15 ans pour la fille ou de plus de 16 ans pour le garçon. De plus en plus, on discute la poursuite du traitement encore quelques années, si le déficit perdure à la fin de l’adolescence.16-18 Le pic de masse osseuse n’étant atteint que dans la troisième décennie, l’intérêt principal de la poursuite du traitement en GH jusqu’à 23-25 ans est de permettre à ces jeunes adultes d’atteindre ce pic de formation osseuse afin d’éviter le développement d’une ostéopénie/ostéoporose de façon prématurée, avec augmentation de deux à trois fois du risque de fracture. Un autre intérêt du traitement est l’effet sur la composition corporelle, le déficit en GH étant associé à une augmentation de la masse grasse et à une diminution de la masse maigre, musculaire en particulier. Cette constellation métabolique paraît associée chez l’adulte à une augmentation des facteurs de risque cardiovasculaires, mais leur correction avec la poursuite du traitement de GH à vie et surtout l’impact sur la mortalité globale n’est actuellement pas prouvée chez l’adulte. Le dernier point pouvant motiver la poursuite de la substitution en GH quelques années après la fin de la croissance serait l’impact de ce déficit sur la qualité de vie. Il existe peu d’outils utilisables pour quantifier la qualité de vie. Jusqu’à présent, chez l’adulte et dans le monde anglophone, il existe un seul questionnaire validé dans l’insuffisance somatotrope (QoL-AGHDA, Quality of Life assessment of GH deficiency in adults). Ce type de questionnaire n’est pas un outil diagnostique, mais permet d’évaluer l’impact du traitement ou de l’arrêt de celui-ci sur la qualité de vie.
La phase de transition
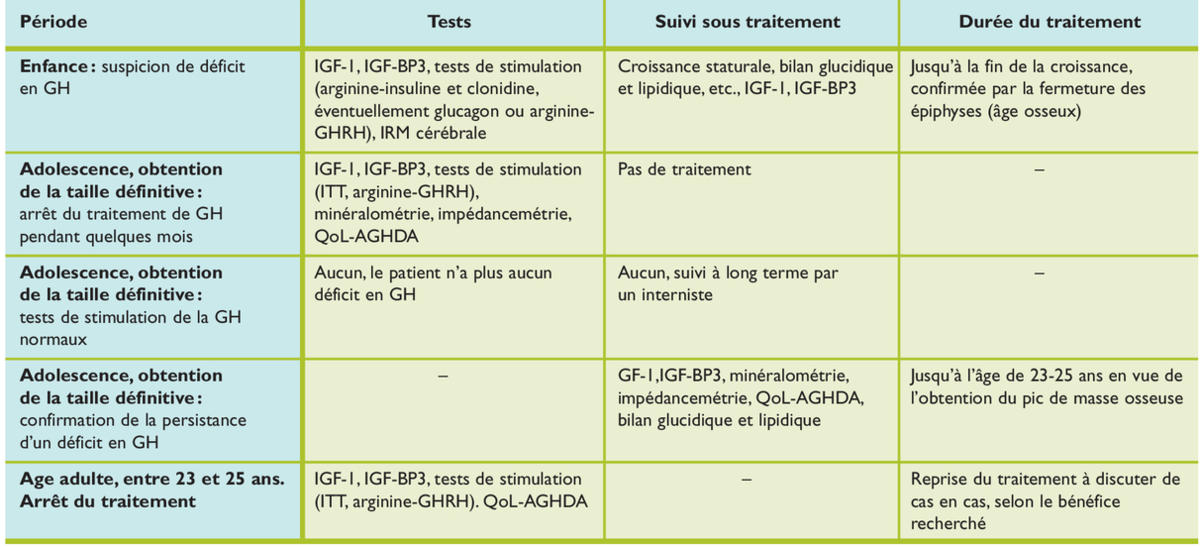
Schéma décisionnel
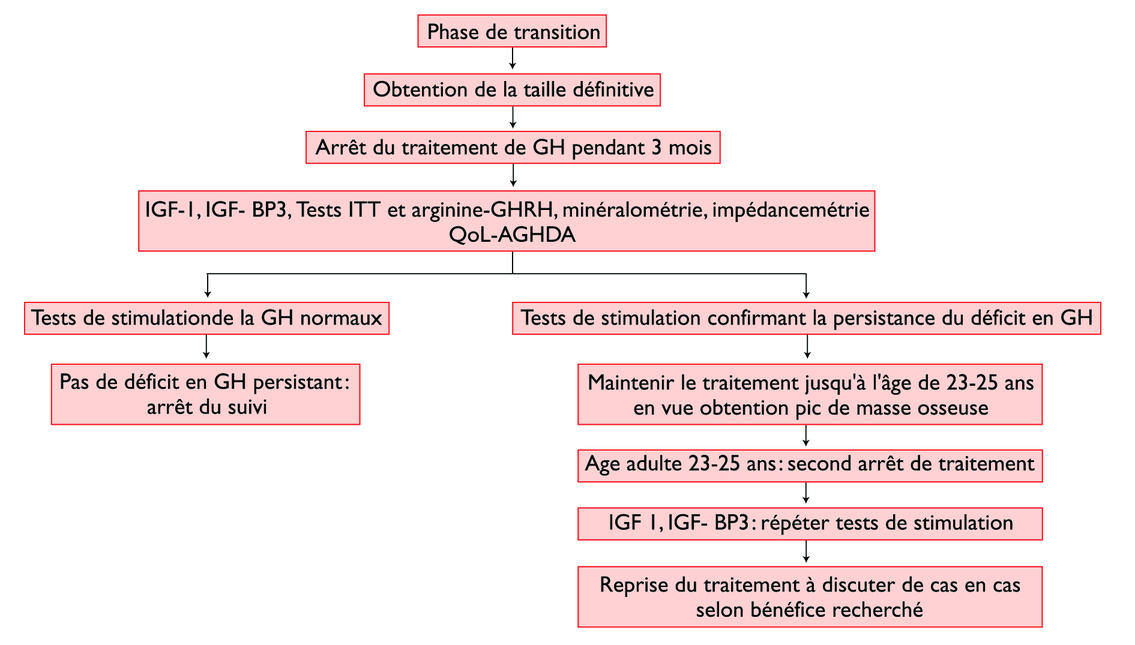
Les coûts des traitements par GH restent très élevés : de l’ordre de Fr. 40 000.-/an chez l’enfant (voire Fr. 80 000.– dans le syndrome de Turner qui nécessite des doses plus élevées), un peu moins à l’âge adulte car les doses sont moindres. Après l’obtention de la taille définitive et la confirmation de la persistance du déficit en GH, chaque cas doit être analysé individuellement en tenant compte des éventuels symptômes liés au déficit en GH. ■
> En cas de suspicion de déficit en hormone de croissance chez l’enfant, des tests de stimulation sont nécessaires en milieu spécialisé
> L’obtention d’une taille dans la taille-cible familiale n’est pas le seul objectif du traitement en GH
> A l’adolescence, une fenêtre thérapeutique doit être instaurée pour effectuer de nouveaux tests de stimulation,de nombreux patients n’étant plus déficients en GH en cas de déficit isolé
> Les enfants ayant subi une irradiation cérébrale ont un déficit en GH définitif et complet, ainsi qu’un pan-hypopituitarisme qui peut survenir jusqu’à une dizaine d’années après l’irradiation
> En cas de déficit persistant, le traitement en GH est souvent continué jusqu’à l’âge de 23-25 ans afin d’atteindre une masse osseuse adéquate
> Le traitement en GH est cher et l’indication à la poursuite du traitement à l’âge adulte (après l’âge de 25 ans) doit être posée par le spécialiste
Le produit a bien été ajouté au panier ! Vous pouvez continuer votre visite ou accéder au panier pour finaliser votre commande.
Veuillez entrer votre adresse email ci-dessous pour recevoir un lien de réinitialisation de mot de passe
Vous pouvez créer votre nouveau mot de passe ici
Certains de ces cookies sont essentiels, tandis que d'autres nous aident à améliorer votre expérience en vous fournissant des informations sur la manière dont le site est utilisé.
Les cookies nécessaires activent la fonctionnalité principale. Le site Web ne peut pas fonctionner correctement sans ces cookies et ne peut être désactivé qu'en modifiant les préférences de votre navigateur.
Ces cookies permettent d’obtenir des statistiques de fréquentation anonymes du site de la Revue Médicale Suisse afin d’optimiser son ergonomie, sa navigation et ses contenus. En désactivant ces cookies, nous ne pourrons pas analyser le trafic du site de la Revue Médicale Suisse
Ces cookies permettent à la Revue Médicale Suisse ou à ses partenaires de vous présenter les publicités les plus pertinentes et les plus adaptées à vos centres d’intérêt en fonction de votre navigation sur le site. En désactivant ces cookies, des publicités sans lien avec vos centres d’intérêt supposés vous seront proposées sur le site.
Ces cookies permettent d’interagir depuis le site de la Revue Médicale Suisse avec les modules sociaux et de partager les contenus du site avec d’autres personnes ou de les informer de votre consultation, lorsque vous cliquez sur les fonctionnalités de partage de Facebook et de Twitter, par exemple. En désactivant ces cookies, vous ne pourrez plus partager les articles de la Revue Médicale Suisse depuis le site de la Revue Médicale Suisse sur les réseaux sociaux.